
Structured Populations of Critically Endangered Yellow Water Lily (Nuphar shimadai Hayata, Nymphaeaceae)
 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Genetic Diversity of Nuphar shimadai Populations in Northern Taiwan
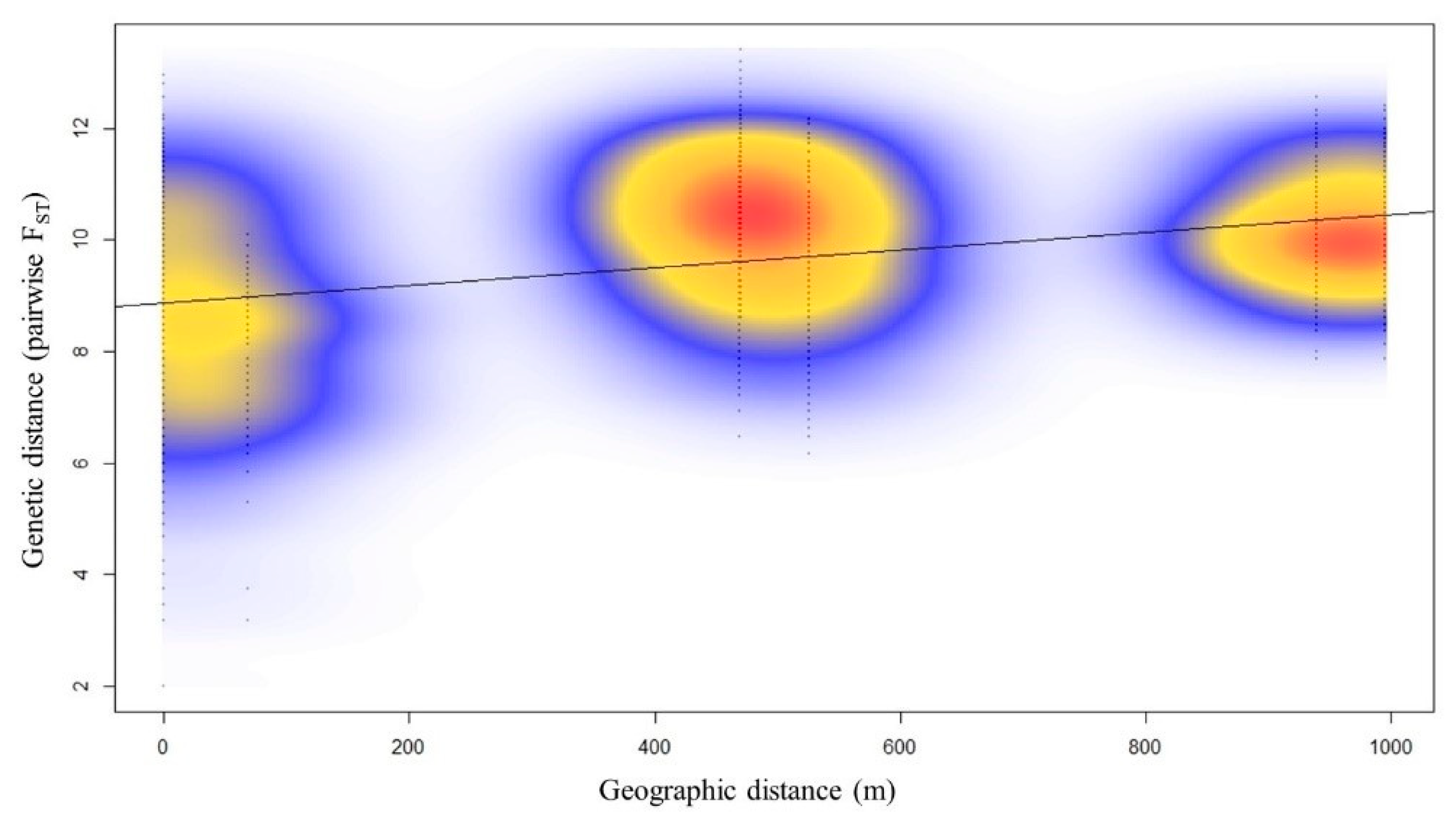
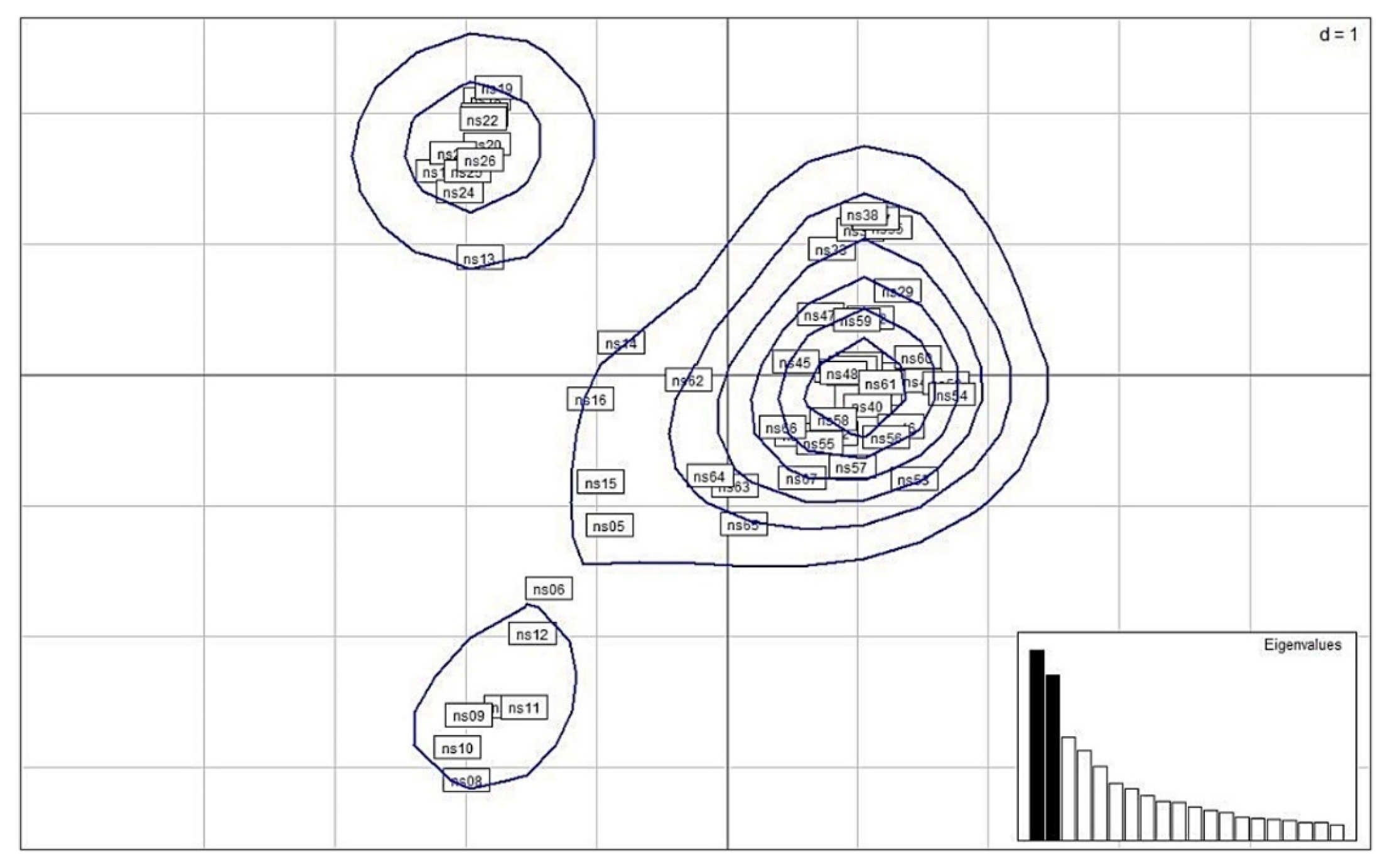
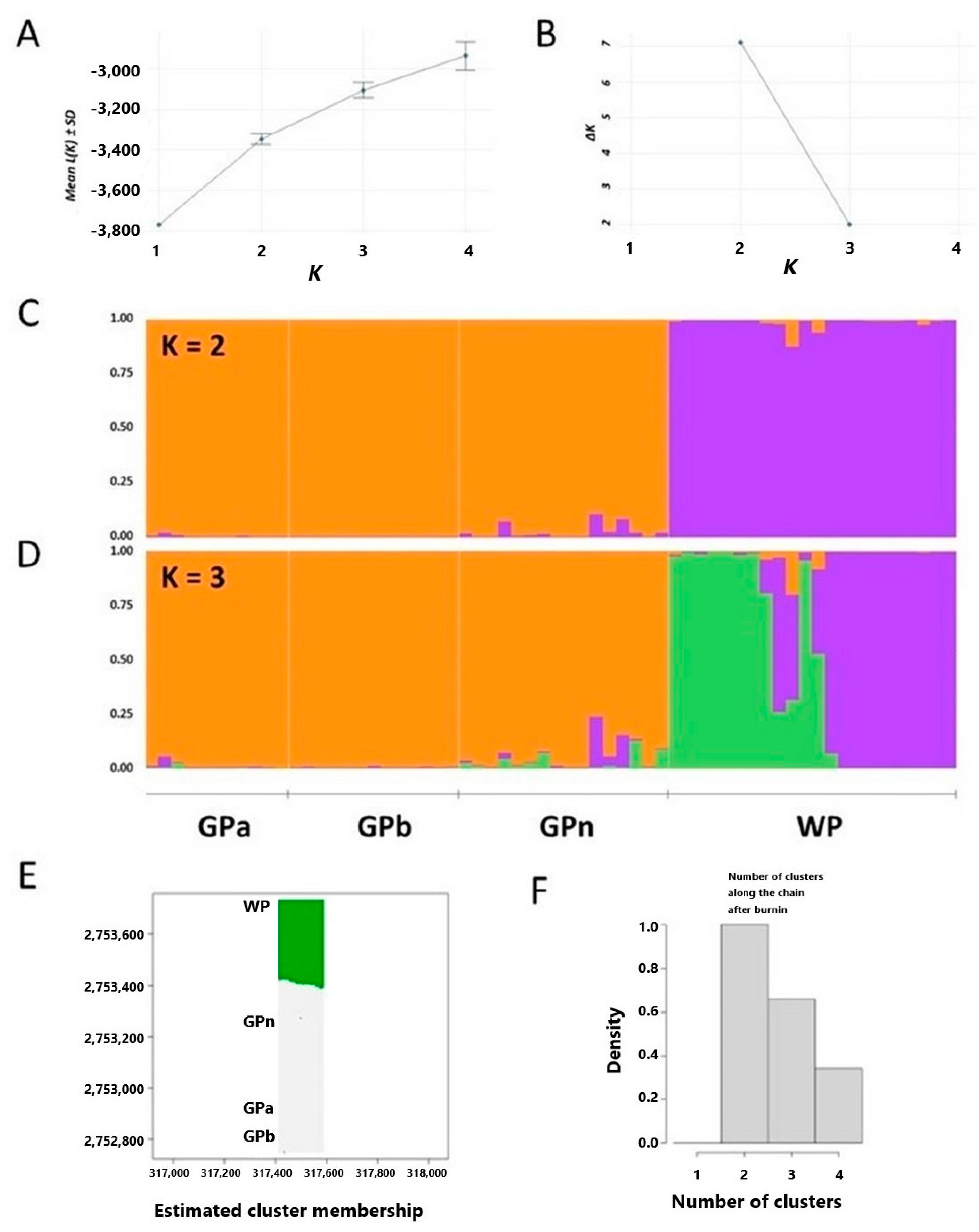
2.2. Population Structure of Nuphar shimadai
3. Discussion
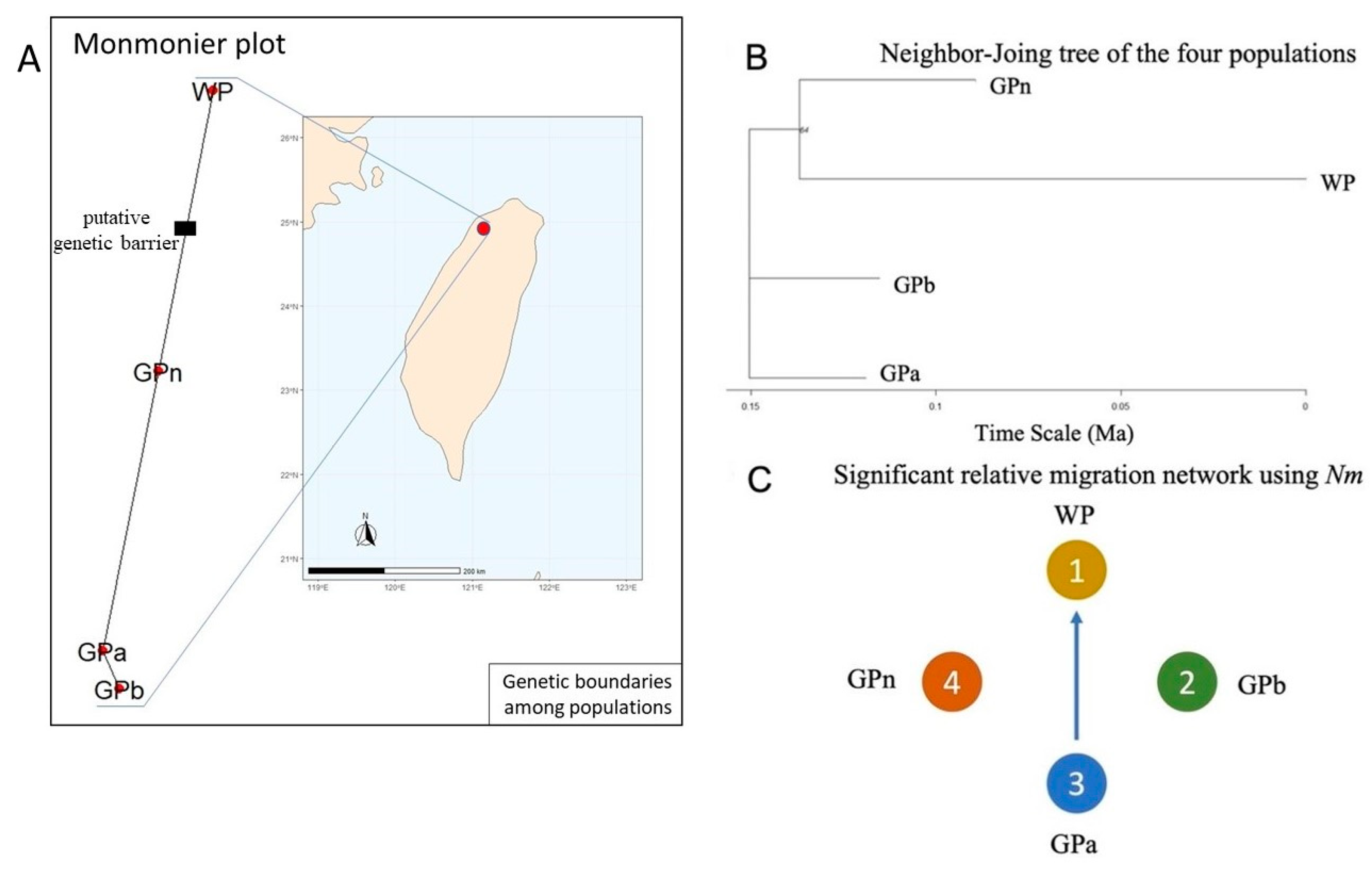
3.1. Genetic Diversity Analysis Revealed GPa Population as the Center of Origin
3.2. Genetic Structure Detected Due to a Genetic Barrier to WP, the Northernmost Population
4. Materials and Methods
4.1. Sampling and DNA Extraction
4.2. SSR Selection and Amplification
4.3. Molecular Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Padgett, D.J. A monograph of Nuphar (Nymphaeaceae). Rhodora 2007, 109, 1–95. Available online: https://www.jstor.org/stable/23314744 (accessed on 12 February 2022). [CrossRef]
- Yang, Y.P.; Lu, S.Y. Nymphaeaceae. In Flora of Taiwan, 2nd ed.; Editorial Committee of the Flora of Taiwan: Taipei, Taiwan, 1995; Volume 2, pp. 610–614. [Google Scholar]
- He, D.; Gichira, A.W.; Li, Z.; Nzei, J.M.; Guo, Y.; Wang, Q.; Chen, J. Intergeneric relationships within the early-diverging angiosperm family Nymphaeaceae based on chloroplast phylogenomics. Int. J. Mol. Sci. 2018, 19, 3780. [Google Scholar] [CrossRef]
- Gruenstaeudl, M. Why the monophyly of Nymphaeaceae currently remains indeterminate: An assessment based on gene-wise plastid phylogenomics. Plant Syst. Evol. 2019, 305, 827–836. [Google Scholar] [CrossRef]
- Global Diversity Information Facility. Nuphar shimadae Hayata. Available online: https://www.gbif.org/occurrence/search?taxon_key=4940220 (accessed on 7 September 2022).
- Editorial Committee of the Red List of Taiwan Plants. The Red List of Vascular Plants of Taiwan; Endemic Species Research Institute, Forestry Bureau, Council of Agriculture, Executive Yuan and Taiwan Society of Plant Systematics: Taipei, Taiwan, 2017; 194p. Available online: https://www.tesri.gov.tw/Uploads/userfile/A6_2/2019-02-25_1315069780.pdf (accessed on 12 February 2022).
- Ouborg, N.J.; Goodall-Copestake, W.P.; Saumitou-Laprade, P.; Bonnin, I.; Epplen, J.T. Novel polymorphic microsatellite loci isolated from the yellow waterlily. Nuphar Lutea Mol. Ecol. 2000, 9, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Watanabe, S.; Shiga, T.; Isagi, Y. Microsatellite markers for Nuphar japonica (Nymphaeaceae), an aquatic plant in the agricultural ecosystem of Japan. Appl. Plant Sci. 2016, 4, 1600082. [Google Scholar] [CrossRef]
- Lu, H.Y.; Shih, H.C.; Ju, L.P.; Hwang, C.C.; Chiang, Y.C. Characterization of 39 microsatellite markers from Nuphar shimadai (Nymphaeaceae) and cross-amplification in two related taxa. Appl. Plant Sci. 2018, 6, e01188. [Google Scholar] [CrossRef]
- Padgett, D.J.; Les, D.H.; Crow, G.E. Phylogenetic relationships in Nuphar (Nymphaeaceae): Evidence from morphology, chloroplast DNA, and nuclear ribosomal DNA. Am. J. Bot. 1999, 86, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Insect Mol. Biol. 2006, 9, 615–629. [Google Scholar] [CrossRef]
- Grünwald, N.J.; Kamvar, Z.N.; Everhart, S.E.; Tabima, J.F.; Knaus, B.J. Population Genetics and Genomics in R; Oregon State University: Corvallis, OR, USA, 2017; Available online: https://grunwaldlab.github.io/Population_Genetics_in_R/ (accessed on 20 June 2022).
- Pavlopoulos, G.A.; Soldatos, T.G.; Barbosa-Silva, A.; Schneider, R. A reference guide for tree analysis and visualization. BioData Min. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D.; Slade, R.; Burden, M.; Estoup, A. Direct, real-time estimation of migration rate using assignment methods: A simulation-based exploration of accuracy and power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Shiga, T.; Yokogawa, M.; Kaneko, S.; Isagi, Y. Genetic diversity and population structure of Nuphar submersa (Nymphaeaceae), a critically endangered aquatic plant endemic to Japan, and implications for its conservation. J. Plant Res. 2017, 130, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, L.; Keenan, K.; Zackrisson, M.; Prodöhl, P.; Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016, 6, 3461–3475. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Hills, H.H. Silica gel: An ideal material for field preservation of leaf samples for DNA studies. Taxon 1991, 40, 215–220. [Google Scholar] [CrossRef]
- Liao, P.C.; Gong, X.; Shih, H.C.; Chiang, Y.C. Isolation and characterization of eleven polymorphic microsatellite loci from an endemic species, Piper polysyphonum (Piperaceae). Conserv. Genet. 2009, 10, 1911–1914. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Shih, H.C.; Chang, L.W.; Li, W.R.; Lin, H.Y.; Ju, L.P. Isolation of 16 polymorphic microsatellite markers from an endangered and endemic species, Podocarpus nakaii (Podocarpaceae). Am. J. Bot. 2011, 98, e306–e309. [Google Scholar] [CrossRef]
- R Core Team. Hierarchical Clustering. The R Stats Package (R 4.1.0). 2021. Available online: https://uc-r.github.io/hc_clustering (accessed on 20 June 2022).
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.K. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using Adegenet 2.1.0; MRC Centre for Outbreak Analysis and Modelling, Imperial College London: London, UK, 2017; 43p. [Google Scholar]
- Jombart, T. A Tutorial for the Spatial Analysis of Principal Components (sPCA) Using Adegenet 2.0.0; MRC Centre for Outbreak Analysis and Modelling, Imperial College London: London, UK, 2015; 50p. [Google Scholar]
- Pritchard, J.K.; Wen, X.; Falush, D. Documentation for Structure Software: Version 2.3; University of Chicago: Chicago, IL, USA, 2010; 38p, Available online: https://web.stanford.edu/group/pritchardlab/structure_software/release_versions/v2.3.4/structure_doc.pdf (accessed on 12 March 2021).
- Pagnotta, M.A. Comparison among methods and statistical software packages to analyze germplasm genetic diversity by means of codominant markers. J (Multidiscip. Sci. J.) 2018, 1, 197–215. [Google Scholar] [CrossRef]
- Guillot, G.; Estoup, A.; Santos, F. Population Genetic and Morphometric Data Analysis Using R and the GENELAND Program. The Geneland Development Group, 2012. Available online: https://backend.orbit.dtu.dk/ws/portalfiles/portal/51230834/Geneland-Doc.pdf (accessed on 12 March 2021).
- Rousset, F. Genepop v. 4.7.5. 2020. Available online: https://kimura.univ-montp2.fr/~rousset/Genepop.htm (accessed on 12 March 2021).
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | Diversity Indices | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Na | Ne | I | Ho | He | uHe | F | %P | |
| WP | 22 | 2.795 | 1.930 | 0.687 | 0.248 | 0.402 | 0.411 | 0.444 | 87.18 |
| GPb | 13 | 1.974 | 1.600 | 0.431 | 0.221 | 0.266 | 0.276 | 0.344 | 61.54 |
| GPa | 11 | 2.077 | 1.554 | 0.444 | 0.228 | 0.271 | 0.284 | 0.412 | 71.79 |
| GPn | 16 | 2.744 | 1.822 | 0.652 | 0.199 | 0.383 | 0.395 | 0.612 | 94.87 |
| Mean | 2.390 | 1.727 | 0.554 | 0.224 | 0.331 | 0.342 | 0.453 | 78.85 | |
| SE | 0.100 | 0.059 | 0.033 | 0.028 | 0.019 | 0.019 | 0.055 | 7.50 | |
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation | p-Value |
|---|---|---|---|---|
| Among populations | 160.56 | 1.4491 | 17.0657 | 0.0000 * |
| Among individuals Within populations | 560.64 | 2.6309 | 30.9835 | 0.0000 * |
| Within individuals | 273.50 | 4.4113 | 51.9508 | 0.0000 * |
| Total | 994.70 | 8.4913 |
| Population | IAM 1-Tailed | IAM 2-Tailed | TPM 1-Tailed | TPM 2-Tailed | SMM 1-Tailed | SMM 2-Tailed | Mode-Shift |
|---|---|---|---|---|---|---|---|
| WP | 0.0000 | 0.0000 | 0.0013 | 0.0027 | 0.1140 | 0.2280 | normal |
| GPb | 0.0006 | 0.0012 | 0.0044 | 0.0087 | 0.0302 | 0.0604 | normal |
| GPa | 0.0226 | 0.0451 | 0.0929 | 0.1858 | 0.5401 | 0.9375 | normal |
| GPn | 0.0055 | 0.0110 | 0.1473 | 0.2947 | 0.6834 | 0.6438 | normal |
| Populations | Single-Sample | Two-Sample | ||
|---|---|---|---|---|
| Ne | 95% CI | Ne | 95% CI | |
| WP | 2.3 | 2.1–2.6 | ||
| GPb | 11.7 | 6.1–26.9 | 1.6 | 1.1–2.1 |
| GPa | Infinite | Infinite | ||
| GPn | 23.1 | 15.4–40.0 | 3.0 | 2.1–4.0 |
| Populations | Sampling Sites | Longitude | Latitude | N |
|---|---|---|---|---|
| WP | Taoyuan, Taiwan | 121°11′39″ E | 24°53′16″ N | 22 |
| GPb | Taoyuan, Taiwan | 121°11′34″ E | 24°52′44″ N | 13 |
| GPa | Taoyuan, Taiwan | 121°11′33″ E | 24°52′46″ N | 11 |
| GPn | Taoyuan, Taiwan | 121°11′36″ E | 24°53′01″ N | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantiquilla, J.A.; Lu, H.-Y.; Shih, H.-C.; Ju, L.-P.; Shiao, M.-S.; Chiang, Y.-C. Structured Populations of Critically Endangered Yellow Water Lily (Nuphar shimadai Hayata, Nymphaeaceae). Plants 2022, 11, 2433. https://doi.org/10.3390/plants11182433
Mantiquilla JA, Lu H-Y, Shih H-C, Ju L-P, Shiao M-S, Chiang Y-C. Structured Populations of Critically Endangered Yellow Water Lily (Nuphar shimadai Hayata, Nymphaeaceae). Plants. 2022; 11(18):2433. https://doi.org/10.3390/plants11182433
Chicago/Turabian StyleMantiquilla, Junaldo A., Hsueh-Yu Lu, Huei-Chuan Shih, Li-Ping Ju, Meng-Shin Shiao, and Yu-Chung Chiang. 2022. "Structured Populations of Critically Endangered Yellow Water Lily (Nuphar shimadai Hayata, Nymphaeaceae)" Plants 11, no. 18: 2433. https://doi.org/10.3390/plants11182433