
Abstract
Viola uliginosa Besser is a European violet having its main distribution range in the Baltic Sea region. Today it is considered endangered and threatened. Species of Violaceae from different genera and sections are known to produce cyclotides, cyclic polypeptides of much interest due to their medicinal properties and chemical structure. The present study introduced a rare species of violet (V. uliginosa) to in vitro culture for biodiversity protection and as a model for cyclotide biosynthesis research in the Violaceae. Leaf and petiole fragments were cultured on MS medium solidified with agar and supplemented with different concentrations of plant growth regulators: TDZ, KIN and 2,4-D. Direct and indirect (via callus) organogenesis was induced on MS supplemented with TDZ (0.5 or 1 mg l−1) or with equal concentrations (2 mg l−1) of KIN and 2,4-D, followed by callus transfer on 1 mg l−1 TDZ. Shoots were rooted on MS with 2 % sucrose and 0.5 mg l−1 IBA and acclimatized. AFLP marker polymorphism was low but flow cytometry revealed that a large share of the obtained regenerants were tetraploid (2C = 4x = 2.7–2.8 pg), unlike the maternal diploid plants (2C = 2x = 1.4 pg). Eleven different cyclotides were distinguished in the aerial parts of maternal plants. Cyclotide production was significantly higher in tetraploid than in diploid plants regenerated in vitro.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Viola uliginosa Besser, the swamp violet, is a European species having its main range of distribution in the Baltic Sea region. The species is declining throughout its range. In many countries (e.g. Poland, Germany, Sweden, Russia) it is considered endangered or even threatened with extinction (Baryła and Kuta 2001; Ingelög et al. 1993; Gärdenfors 2005; Korneck et al. 1996; Kotiranta et al. 1998; Ryttäri and Kettunen 1997; Stoltze and Pihl 1998). Viola uliginosa was originally described in 1809 by Besser from Rząska near Cracow, Poland (Locus classicus), and was thereafter identified at several localities in southern Poland (Baryła and Kuta 2001; Krawczyk et al. 2008). The high genetic uniformity of the locus classicus population, due to clonal propagation and population shrinkage, is a factor in its endangerment (Cieślak et al. 2006).
Culture of plants in vitro is increasingly used for biodiversity conservation, especially for rare endemic species, and is also an important component of plant genetic resource management (Pence 2013 and references therein; Sarasan et al. 2006; Thorpe 2007). Somaclonal variation induced by culture conditions limits the use of in vitro micropropagation, as it leads to changes in nuclear and cytoplasmic genomes and causes genetic differentiation of regenerated plants (D’Amato 1991; Larkin and Scowcroft 1981; Lee and Phillips 1988; Neelakandan and Wang 2012; Rodriguez-Enriquez et al. 2011; Vazquez 2001; Wang and Wang 2012). Confirmation of the genetic identity with maternal material, and the genome size and reproductive normality of regenerated plants, is required before reintroduction (Nybom et al. 2014; Ochatt et al. 2011; Singh et al. 2013; Thiem et al. 2013; Thiem and Sliwinska 2003). Molecular markers are valuable tools for detecting the sequence variation of closely related genomes such as those between source plants and somaclones regenerated through tissue culture. Molecular marker systems—random amplified polymorphic DNA (RAPD), inter-simple sequence repeat (ISSR), simple sequence repeat (SSR), amplified fragment length polymorphism (AFLP) and restriction fragment length polymorphism (RFLP)—each having its advantages and limitations, are used to estimate somaclonal variation in tissue culture (Bairu et al. 2011; Nybom et al. 2014; Rewers et al. 2012; Singh et al. 2013). Flow cytometry for somaclone genome size assessment can be used to complement molecular marker analyses (Bairu et al. 2011; Ochatt et al. 2011; Rewers et al. 2012).
On the other hand, somaclonal variation can lead to new intraspecific forms with unique traits affecting the quality and quantity of the primary and secondary metabolites they produce. The most valuable forms can then be selected and used to establish large-scale culture systems (suspension cell culture or organ culture in bioreactors, or as regenerated plants) for production of those compounds (Ahmad et al. 2013; Bourgaud et al. 2001; Rout et al. 2000).
Plants of the Violaceae family are known for their expression of cyclotides (Burman et al. 2014). The chemistry and biology of this unique family of ribosomally produced plant peptides are of interest to numerous researchers (Arnison et al. 2013). Cyclotides have a head-to-tail cyclic peptide backbone and three disulfide bonds arranged in a cystine knot (Craik et al. 1999; Rosengren et al. 2003). They are expressed as complex peptide mixtures in apparently all species of Violaceae; outside of that plant family, cyclotides or cyclotide-like proteins or genes have been found in Rubiaceae, Cucurbitaceae, Fabaceae, Solanaceae and Poaceae (Burman et al. 2014; Gerlach et al. 2013; Nguyen et al. 2013; Poth et al. 2012). Numerous biological activities have been reported for cyclotides, including uterotonic, hemolytic, inhibition of neurotensin action, anti-HIV and cytotoxic (Burman et al. 2014). They are active against different bacteria and insect larvae (Jennings et al. 2005 Pränting et al. 2010). The insecticidal and antimicrobial activities suggest their role as components of the plant defense system (Craik 2012).
The unique structure of cyclotides makes chemical synthesis difficult; the most widely used method of obtaining cyclotides in large amounts still is extraction from plant material (Craik and Conibear 2011; Dörnenburg 2010). This requires large amounts of plant material, as the content of cyclotides in tissues is low and their availability may be limited, especially in the case of rare species. In vitro methods for cyclotide production were developed recently but the study was limited to Oldenlandia affinis (Rubiaceae) (Dörnenburg 2010).
In this study we apply plant cell micropropagation in vitro for conservation of the rare violet species, V. uliginosa and as a model for cyclotide research in the Violaceae. We developed a protocol for in vitro micropropagation of V. uliginosa, detected somaclonal variation using AFLP molecular markers, and estimated genome size in mother plants and the ploidy level of clones derived from tissue culture. We discuss in vitro culture schemes for cyclotide production and for somaclonal variation-enhanced cyclotide biosynthesis.
Materials and methods
Plant material
Plants from natural site
Plants of V. uliginosa were collected in May 2010 in Nowa Dęba (southern Poland; N50°25′14,90″, E21°47′08,72″, alt 171.7 m) under a permit from the Regional Directorate for Environmental Protection and were identified by E. Kuta. Only 4 or 5 plants were harvested from the population and cultivated in pots under natural light at room temperature in the Department of Plant Cytology and Embryology, Institute of Botany, Jagiellonian University, Cracow.
Somaclones from the initial screening experiment
Acclimatized regenerants obtained in the initial screening experiment, grown in an experimental field in Modlnica near Cracow, were collected and included in genetic analyses of variability (AFLP) and genome size/ploidy level estimates. All plants were obtained from leaf explants in several steps of prolonged indirect organogenesis to increase somaclonal variation and the frequency of polyploid regenerants. The initial screening experiment tested different explant types (stigma, cotyledon, hypocotyl, sepal, petal, peduncle, petiole, leaf) and media compositions. Callus proliferation was maintained for 6 months on agar-solidified MS (Murashige and Skoog 1962) medium supplemented with 2,4-D (2 mg l−1) or KIN (2 mg l−1) and then shoot proliferation was induced by two-step transfer on MS + 1 mg l−1 KIN or 1 mg l−1 KIN + 0.03 mg l−1 2,4-D and then on MS enriched with TDZ (0.5 mg l−1 or 1.0 mg l−1). The shoots were rooted on MS medium + 2 % sucrose + IBA (0.5 mg l−1) solidified with agar. Rooted shoots were acclimatized to a sterilized mixture of commercially available soil and perlite (1:1) in a minigreenhouse for 1–2 months and then planted in the experimental plot in Modlnica.
In vitro culture
Sterilization
Petiole and leaves were surface-sterilized by soaking with 70 % ethanol for 60 s, then in ACE commercial bleach diluted with distilled water (1:4) for 12 min, and finally rinsed three times (5 min each) in sterile distilled water. After sterilization the leaves were cut into approx. 0.5 cm × 0.5 cm fragments, and petioles into 0.3–0.5 cm long fragments. Culture media and tools were autoclaved in a steam autoclave (121 °C, pressure 1.05 bar) for 30 min.
Media composition and treatments
Explants were cultured on MS basal medium supplemented with different combinations and concentrations of plant growth regulators (KIN, 2,4-D, TDZ, IBA, all from Sigma-Aldrich) solidified with 4 g l−1 agar (MP Biomedicals) and supplemented with 30 g l−1 sucrose.
Prior to autoclaving, all media were adjusted to pH 5.7–5.8 using 0.1 N HCl/NaOH. For callus tissue induction, media supplemented with 2 mg l−1 KIN + 2 mg l−1 2,4-D were used. After 2 months of callusing the explants were transferred to media containing 1 or 0.5 mg l−1 TDZ for organogenesis induction. For direct organogenesis both explant types were cultured on 1 or 0.5 mg l−1 TDZ. Sterile leaf and petiole fragments (initial explants) were placed in medium-filled Petri dishes (10–15 explants per dish) and sealed with Parafilm®. All experiments were done in three replicates.
Rooting and acclimatization
Shoots with at least two properly formed leaves were separated from the explants and cultured in 50 ml tubes on solid MS basal medium supplemented with 0.5 mg l−1 IBA and reduced sucrose concentration (2 % w/v). Shoots were cultured on the rooting media for up to 3 months. Well-developed plantlets with 2 or 3 roots were acclimatized to an autoclaved mixture of commercially available soil and perlite (1:1) in a minigreenhouse for 1–2 months. Then the plantlets were transferred to soil in flowerpots and under natural light at room temperature. Finally, hardened plants were transferred to the experimental field in Modlnica.
Culture maintenance
Cultures were maintained in a culture room at constant 25 ± 3 °C under a 16 h photoperiod (60–90 µmol m−2 s−1 flux, cool white fluorescent light). All cultures were subcultured every month.
Data analysis
The percentages of explants with callus tissue proliferation/explants with organogenesis were recorded and the effect of explant type was assessed by applying Fisher’s test (two-sided p = 0.05) to the corresponding contingency tables.
Histology
To analyze organogenesis type (direct, indirect) and callus structure, material from culture was fixed in 10 % glutaraldehyde for 24 h, rinsed four times in PBS buffer (pH 7.2), dehydrated in a graded ethanol series (15 min each: 10, 30, 50, 70, 96 %) and kept overnight in absolute ethanol. Then the fixed samples were embedded in Technovit 7100 (2-hydroxyethyl-methacrylate) (Heraeus Kulzer), infiltrated in a mixture of absolute ethanol and Technovit (1 h each: 3:1, 1:1, 1:3 v/v) and stored for 12 h in pure Technovit. Embedded tissues were sectioned 5 µm thick with a rotary microtome (Microm, Adamas Instrumenten), stained with toluidine blue and mounted in Entellan (Merck). Microscopy sections were photographed with a Nikon DS-Fi2 digital camera using NIS-Elements D software.
Flow cytometry
Endopolyploidy determination
Young leaves, petioles (explants for in vitro culture) and different callus types (morphogenic, amorphous) derived from leaf explants were used. Callus samples were harvested after 1 or 2 passages after transfer of callus tissue to organogenesis medium. Samples were prepared as previously described (Thiem and Sliwinska 2003), using nuclei isolation buffer (200 mM Tris–Cl, pH 7.5, 4 mM MgCl21 × 6 H2O, 0.5 % v/v Triton X-100) supplemented with 1.5 % (w/v) PVP-10 (Jedrzejczyk and Sliwinska 2010) and 4′,6-diamidino-2-phenylindole (2 µg ml−1 DAPI) for DNA staining. Five biological replicates were analyzed with a Partec CCA flow cytometer (Partec GmbH, Münster, Germany) equipped with an HBO lamp, using logarithmic amplification. The DAPI fluorescence of 3,000–7,000 nuclei of each sample was analyzed. The proportion of nuclei with different DNA content, the (4C + 8C + 16C)/2C ratio, and the mean C-value (mean ploidy; Lemontey et al. 2000) were established. Nuclei having at least 8C DNA content were considered to be endopolyploid.
Genome size/ploidy estimation
Leaf samples of mother plants and in vitro regenerated plantlets (originating from initial screening and the present experiments) were prepared as for endopolyploidy estimation, but DAPI was replaced by propidium iodide (50 µg ml−1 PI) and ribonuclease A (50 µg ml−1). Solanum lycopersicum cv. Stupicke (2C = 1.96 pg; Doležel et al. 2007) was the internal standard. PI fluorescence was measured in at least 5,000 nuclei of each sample with a CyFlow SL Green flow cytometer (Partec GmbH, Münster, Germany) equipped with a high-grade solid-state laser with green emission at 532 nm and side (SSC) and forward (FSC) scatter. Analyses used linear amplification. The coefficient of variation (CV) of the G0/G1 peak of Viola ranged from 2.66 to 6.98 %. Nuclear DNA content was calculated using the linear relationship between the ratio of the 2C peak positions of Viola/Solanum on a histogram of fluorescence intensities.
Data analysis
Histograms were evaluated using DPAC v. 2.2 (Partec GmbH, Münster, Germany) for endopolyploidy assessment and FloMax software (Partec GmbH, Münster, Germany) for genome size estimation. The mean (4C + 8C + 16C)/2C ratio, mean C-value (mean ploidy) and mean genome size for explants, calluses and regenerants were statistically analyzed by one-way ANOVA for independent samples and the t test (p = 0.05).
Amplified fragment length polymorphism (AFLP)
DNA isolation
Two or three leaves were collected from mother plants and regenerants, placed in hermetic tubes filled with silica gel (sodium silicate, Sigma) for desiccation, and stored until use. DNA was isolated from approx. 15 mg dried plant material. Extraction was done with the Qiagen Plant DNeasy Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. DNA quality and concentration was roughly estimated by agarose gel electrophoresis and ethidium bromide staining. Extracted DNA was stored at −20 °C prior to analysis.
AFLP procedures
Analyses followed the AFLP protocol (Vos et al. 1995) as modified by Ronikier et al. (2008). EcoRI and MseI were used for restriction digestion of genomic DNA. Selective amplification was performed using three different primer pairs selected after screening for polymorphism, clarity and reproducibility: E-AAC + M-CAT, E-AGG + M-CGT, E-ATT + M-CAT (EcoRI primers were 5′-labeled with fluorescent dye 6-FAM). Fragments were separated by electrophoresis on an ABI Prism 3130 DNA sequencer (Applied Biosystems, Foster City, CA, USA) using POP-7™ polymer, a 36 cm capillary array and GeneScan™500 Rox™ as internal lane size marker. Eight samples were duplicated from single DNA isolates and analyzed independently to test marker reliability. To confirm the results from the first analysis we ran another one under the same conditions.
Data analysis
AFLP data files were aligned using the internal standard and manually scored in Genographer 2.1 (http://sourceforge.net/projects/genographer). Dominant AFLP markers were scored in the 50–500 bp size range and coded as present (1) or absent (0) in a binary data matrix. Only markers unambiguously scoreable (well separated) and repeatable among duplicates were considered in scoring. The error rate was calculated as the share of mismatches in the scoring of AFLP profiles of replicated individuals.
Genetic diversity was quantified as the percentage of polymorphic bands calculated from the analysis. PCoA based on Euclidean distances was used to assess the genetic relationships among individuals. It was calculated and visualized using MVSP software (Kovach Computing Services, Anglesey, Wales, UK). Number of clones was analyzed using R-script AFLPdat (Ehrich 2006; R Development Core Team 2010). Diversity of clones was estimated by applying a moving mismatch threshold allowed among genotypes.
Cyclotide expression
Plant material and preparation of extracts
Viola uliginosa somaclones were cultivated in commercially available soil on a shelf in the laboratory corridor at 20–25 °C under a 16 h photoperiod (70–100 µM photons m−1s−2) for at least 3 months to standardize environmental factors affecting secondary metabolite production.
Aerial parts of mother plants from the pots and regenerants (similar in size) were harvested and freeze-dried. The dry plant material (30–50 mg) was extracted three times using 4 ml 30 % acetonitrile (ACN) and 0.1 % formic acid (FA) in water for 4–12 h each time (24 h total extraction time). The extracts were diluted with 0.1 % FA in water to a final concentration of 10 % ACN in 0.1 % FA, loaded on SPE columns [ISOLUTE C18(EC), 10 ml, 500 mg], washed with 10 % ACN in 0.1 % FA and eluted with 5 ml 60 % ACN, 0.1 % FA. The cyclotides are thus enriched in the 60 % ACN fraction. That fraction was collected and freeze-dried. Samples were dissolved in 10 % ACN and 0.1 % FA in water using 8 µl mg−1 d.w. starting material for extraction. Finally, 10 µl of each sample solution was diluted with 90 µl 10 % ACN 0.1 % FA in water containing the cyclotide kalata B2 as internal standard (500 µg cyclotide/ml, before dilution).
Identification of peptides as cyclotides
The methods used for cyclotide isolation, HPLC purification, cysteine reduction and alkylation as well as trypsin and/or Glu-C cleavage as criteria for cyclotide identification were as described by Gruber et al. (2008).
Liquid chromatography/Mass spectrometry (LC/MS)
Samples were analyzed using ultra performance liquid chromatography coupled to quadropole time of flight mass spectrometry (nanoAcquity UPLC/QTof Micro; Waters, Milford, MA). Samples were separated on reverse phase using a gradient of ACN in FA (0–100 % ACN), and cyclotides were detected in the positive ion mode. A nanoLC column [Waters BEH, 75 µM (i.d) × 150 mm] operated at 0.3 µl/min flow rate was used, combined with nanoelectrospray ionization operated at 4,300 V. Doubly and triply charged cyclotide ions were analyzed, using a mass window of 500–2,000 m/z.
Data analysis
Mass chromatograms were generated using a mass window of 1 Da for the analyzed cyclotides (m/z 3+ ions of 1,042, 1,052 and 1,076 respectively), integrated in Waters MassLynx™ software. Integration was performed with noise peak-to-peak amplitude of 2,000, two smooths (mean smoothing method) with ±3 window size, peak detection with peak tailing reduction until the tailing edge is no more than 50 % wider than the leading edge, and baseline rise by no more than 5 % of peak height. Area under the curve (AUC) was obtained as the relative concentration measure. All AUC values were adjusted to the internal standard (adjustment coefficient = AUCint. standard/AUCint. standard in the sample). Mean AUC ± SD was compared between 2x and 4x plants and the significance of differences between means was tested by one-way ANOVA for independent samples and the t test (p = 0.05).
Results
Micropropagation and acclimatization
Callus tissue and shoot tip formation
After one month of culture on MS + 2 mg l−1 KIN + 2 mg l−1 2,4-D callus induction medium, yellowish amorphous callus tissue proliferated on the cut edges of petiole explants and on the cut edges and surface of leaf explants (Fig. 1a). At 2 months of culture the response rate for both explant types was high, reaching 84.9 % for petiole and 73.4 % for leaf (Table 1). Callus was composed of parenchymatous cells differing in size and vacuolation, as seen in callus sections (Fig. 1g). Following transfer to organogenesis induction media (MS + 1 mg l−1 TDZ), the shoot proliferation response rate was very low for callus derived from both types of explants. Spots of green morphogenic callus tissue (Fig. 1b) developed on only a few explants 3–4 months after transfer. At 6 months after transfer only 6.0 % of the petiole explants and 10.5 % of the leaf explants formed single or multiple shoots (Table 1, Fig. 1c).

Micropropagation, rooting and acclimatization of V. uliginosa. a Nonmorphogenic yellowish callus induced on leaf fragment on MS + 2 mg l−1 KIN + 2 mg l−1 2,4-D after 1 month of culture. b Morphogenic green callus produced after transfer of leaf explants with callus from MS + 2 mg l−1 KIN + 2 mg l−1 2,4-D to MS + 1 mg l−1 TDZ, 3 months on MS with TDZ. c Multiple shoots forming on leaf-derived callus 4 months after transfer to MS + 1 mg l−1 TDZ. d Leaf-derived callus after 3 months of culture on MS + 1 mg l−1 TDZ. e Shoot tips forming on petiole-derived callus after 3 months of culture on MS + 1 mg l−1 TDZ. f Shoot forming directly on petiole fragment after 2 weeks of culture on MS + 0.5 mg l−1 TDZ. g Transverse section of callus tissue proliferating from cut edge of petiole after two weeks of culture on MS + 2 mg l−1 KIN + 2 mg l−1 2,4-D. h, i Longitudinal sections of adventitious shoots forming directly on petiole fragment after 2 weeks of culture on MS + 0.5 mg l−1 TDZ (h) and from callus on leaf fragment after 3 months of culture on MS + 1 mg l−1 TDZ (i). j Regenerated plantlet rooted on 1/2 MS + 0.5 mg l−1 IBA. k Regenerated plant in plastic pot with horticultural substrate. l Regenerated plants grown at experimental field in Modlnica. m Regenerated plant developing chasmogamous flowers
Both explant types formed green organogenic callus on medium supplemented with 1 mg l−1 TDZ. It formed after 2 weeks of culture, and by 3 months of culture the majority of explants (65.6 % petiole explants vs 86.5 % leaf explants, p < 0.05) formed shoot tips at the surface (Table 1, Fig. 1d, e).
Direct organogenesis was obtained only for petiole explants cultured on MS + 0.5 mg l−1 TDZ. Shoot tips formed after 2 weeks of culture at the cut edges of explants (Fig. 1f). At 2 months of culture, 44.1 % of the explants formed 1 or 2 shoots (Table 1).
Direct and indirect organogenesis was confirmed by histological analysis which showed that the shoots developed directly from meristematic centers forming in petiole subepidermal parenchyma on MS + 0.5 mg l−1 TDZ (Fig. 1h), and from callus on leaf fragments on MS + 1 mg l−1 TDZ (Fig. 1i).
Rooting and acclimatization of micropropagated plants
For rooting we used 88 adventitious shoots developed on leaf and petiole explants. Roots formed after 1–3 months of culture (Fig. 1j). The rooting response ratio of explants originating from different treatments ranged from 46.2 to 72.2 % (Table 2). Shoots derived from leaf explants tended to form roots more frequently but the difference was not significant (p > 0.05). Well-developed, hardened plants (Fig. 1k) were transferred to the experimental field (Fig. 1l) in spring. Some of these plants did blossom with chasmogamous and/or semi-cleistogamous flowers in the following months (Fig. 1m). Overall, 50.4 % of the plantlets survived acclimatization (Table 2).
Flow cytometry
Endopolyploidy of explant and callus types
All analyzed samples were polysomatic, comprising nuclei with 2C, 4C, 8C and 16C (the latter only in calluses; Table 3). The mean C-value and (4C + 8C + 16C)/2C were lowest in leaf (2.30 ± 0.02) and highest in brown amorphous callus obtained on medium supplemented with 2 mg l−1 KIN + 2 mg l−1 2,4 D (3.98 ± 0.15). In leaf, 85.60 % of the nuclei showed 2C DNA content (G0/G1 phase of cell cycle), and the share of endopolyploid nuclei (8C) was 0.22 %. Endoreduplication was higher in petiole (10.68 % 8C nuclei) than in leaf (mean C-values for petiole vs leaf, p < 0.05, Table 3). In all callus types the share of 2C nuclei was lower than in the explant (mean C-value vs leaf, p < 0.05), and in two of the three types the second endocycle occurred, revealed in the presence of 16C nuclei (Table 3). Organogenesis induction lowered the mean C-value significantly.
Genome size and ploidy of regenerated plants
The genome size of diploid (2n = 2x = 20) mother plants was 1.37 pg/2C (Table 4). To our knowledge this is the first report on genome size in V. uliginosa. Among the regenerants, tetraploid plants (2C = 2.7 pg) occurred in addition to diploids. The share of tetraploids was highest (70.6 %) in plants obtained via indirect organogenesis with a morphogenesis induction step (long culture in initial screening experiment). Shorter culture on MS medium supplemented with 1 mg l−1 TDZ, alone or after transfer from KIN and 2,4-D, resulted in regeneration of 25.0–35.7 % tetraploids. All the plants from direct organogenesis on MS medium supplemented with 0.5 mg l−1 TDZ were diploids. None of the mean genome size differences among diploid and tetraploid plants obtained from the different culture variants were significant, nor those between mother plants and diploid regenerants (p > 0.05).
Genetic differentiation of regenerated plants estimated by AFLP markers
Fifty-three samples were analyzed: 51 regenerated plants obtained by micropropagation (27 from the initial screening experiment, 24 from the present experiment) and 2 mother plants. Genetic variability within and between all groups was low, and the samples differed from each other at only a few markers. Overall, only 20 (17.09 %) of the 117 amplified bands were polymorphic.
Four groups were distinguished in PCoA (Fig. 2). Most of the somaclones formed one large group (C1/N = 33) with maternal plant (ND5). A few other samples formed a very closely related cluster (C3/N = 7), with another maternal plant genotype (ND3). Altogether, 78.4 % of the clones grouped with their maternal plants. Two other groups included the few remaining plants (C2/N = 9 and C4/n = 2), with only the smallest group C4 differing significantly and accounting for a large part of total polymorphism. Only clones from the latter group differed from the others by more than five AFLP markers. Within groups the differences between genotypes (clones) were minor and did not exceed two markers except in C2, the most polymorphic group, where they differed by three markers. All the somaclones in groups C2 and C4 were obtained in the initial screening experiment via indirect organogenesis with a morphogenesis induction step, which consisted consecutive callus passages.

Principal coordinate analysis (PCoA) of 2 maternal and 51 regenerated V. uliginosa plants based on Euclidean distances. C1–C4, groups of somaclones; ND3, ND5, maternal plants
Cyclotide biosynthesis in regenerated plants
Eleven different compounds with molecular weights ranging from 2,876 to 3,273 Da were detected by liquid chromatography/mass spectrometry (LC–MS) in the aerial parts of V. uliginosa mother plants (Online Resource 1). After reduction and alkylation, the differences in mass corresponded to the presence of three disulfide bonds, and Glu-C cleavage resulted in one linear fragment (mass difference corresponding to one H2O), fulfilling the criteria described by Gruber et al. (2008) to classify them as cyclotides.
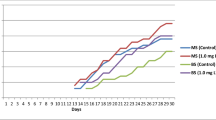
Fifteen regenerated plants, including 9 diploids (2x) and 6 tetraploids (4x) were suitable for analysis of relative cyclotide biosynthesis. In the cyclotide part of the mass chromatograms the MS spectra of the somaclones and mother plants were similar; that is, no additional cyclotides were found and no cyclotides were missing. Mean AUC difference analysis for the three most abundant cyclotides favored the 4x plants for all analyzed peaks, with the biggest difference in means for the 3,123 Da cyclotide (significant at p = 0.01) (Fig. 3). The difference in mean AUC for the 3,225 Da and 3,152 Da cyclotides was not statistically significant due to high standard deviations, as some somaclones showed high cyclotide accumulation independent of ploidy level. The mother plants, at a different development stage and cultivated in different conditions, showed nearly half the AUC of any of the 2x or 4x regenerated plants (Online resource 2).

Relative concentration (in terms of AUC) of three main cyclotides (3,123, 3,225, 3,152 Da) in extracts from 4x and 2x regenerated V. uliginosa plants. Bars and values in brackets indicate standard deviation (SD). N, number of biological replicates
Discussion
Violaceae have rarely been micropropagated in vitro; it has been done in two Hybanthus species (Bidwell et al. 2001; Prakash et al. 1999), V. patrinii (Chalageri and Babu 2012), V. pilosa (Soni and Kaur 2013) and V. wittrockiana (Wang and Bao 2007). Here we applied in vitro micropropagation to the rare species V. uliginosa. The method was effective, yielding 61 regenerated plantlets. Most of the regenerants were produced by indirect (via callus) organogenesis, as in other studies on in vitro micropropagation of Violaceae (Bidwell et al. 2001; Chalageri and Babu 2012; Prakash et al. 1999; Soni and Kaur 2013; Wang and Bao 2007). A higher TDZ concentration (1 mg l−1) was needed for induction of caulogenesis. Naeem et al. (2013) and Wang and Bao (2007) noted the importance of TDZ for culture of V. odorata and V. wittrockiana, respectively. In our study we showed that a lower concentration of TDZ (0.5 mg l−1) can be used to induce direct organogenesis from petiole explants.
The adequate explant types, in terms of availability and efectiveness of callogenesis were selected in the initial screening experiment, namely the petiole and leaf. In the present study nonmorphogenic callus proliferation on MS + 2 mg l−1 KIN + 2 mg l−1 2,4-D and morphogenic callus and shoot formation (after addition of 1 mg l−1 TDZ) were more efficient when leaf explant was used, because the whole explant surface was involved. Moreover, leaf contained fewer endopolyploid cells, a potential source of polyploid regenerants. Most of other studies on micropropagation of species from the Violaceae used only petiole (Chalageri and Babu 2012, Wang and Bao 2007), bud (Soni and Kaur 2013) or seed (Prakash et al. 1999) explants. Only in one case leaf explants were compared with stem (Bidwell et al. 2001). Comparing the two explant types, our study indicates that leaf fragments are more suitable than petioles for in vitro cultures and in vitro micropropagation of V. uliginosa.
Somaclonal variation induced by culture conditions makes it essential to confirm the genetic uniformity of regenerated plants with the mother plant(s) before reintroduction to natural sites or planting in botanic gardens. In our study, marker polymorphism was generally low: 78.4 % of the clones grouped with their maternal plants and the differences were limited to single markers. Low levels of marker polymorphism have been described also in studies on other Violaceae species, using different molecular fingerprinting techniques—RAPD or ISSR (Chalageri and Babu 2012; Soni and Kaur 2013). Nevertheless, the genetic uniformity of the regenerated plants with the mother plants, as detected by AFLP analyses, clearly depended on the type of organogenesis. Shoot formation on media with TDZ at both doses (1 and 0.5 mg l−1) gave lower marker polymorphism in comparison with long-term culture of nondifferentiated callus and indirect shoot formation. The only two groups of somaclones differing from their mother plants and accounting for a large part of total polymorphism were obtained via long-term indirect organogenesis (somaclones from the initial screening experiment study). This suggests that more direct methods should be preferred for micropropagation purposes, even when the process is less effective.
The genetic stability of regenerated plants as estimated by molecular markers is not equivalent to ploidy (autopolyploidy) level, though. Molecular markers can identify gene polymorphism and are useful for exploring polyploid (mainly allopolyploid) genomes in plants, but identifying autopolyploids (produced via somaclonal variation in culture) by these methods is problematic (Aversano et al. 2012). The value of flow cytometry technique for somaclone genome size assessment has been demonstrated by a number of authors (e.g., Bairu et al. 2011; Ochatt et al. 2011). In our study, ~24 % of the regenerants from direct and indirect organogensis (on MS media supplemented with TDZ, 2 mg l−1 KIN + 2 mg l−1 2,4-D transferred to 1 mg l−1 TDZ) represented tetraploid level, regardless of their low genetic variability. The frequency of tetraploid plants dramatically increased (to approx. 70 %) when regeneration was via long-term indirect organogenesis. This was probably a consequence of the higher endopolyploidy (reaching even 16C) of the long-term callus tissue from which adventitious shoots developed. Studies on endoploidy level in dedifferentiated callus support that suggestion (Thiem and Sliwinska 2003). Taking the abovementioned into consideration it seems that molecular techniques like AFLP may not be sufficient to detect somaclonal variation and assessment of ploidy level of regenerated plants should be a standard procedure before reintroduction, especially in case of indirect organogenesis.
Somaclonal variation limits the usefulness of micropropagation for conservation of rare and/or endangered plants, as it leads to genetic differentiation and changes in the ploidy level of regenerated plants, but it can improve the economic value of some species, especially those producing valuable metabolites (Bouque et al. 1998; Kulkarni et al. 2012). Cyclotides originate from ribosomal synthesis and are encoded in the nuclear genome (Craik and Conibear 2011). Therefore, mutations occurring in culture can result in higher production or new structures with new properties. For example, 12 novel cyclotides not present in the wild plant were found in O. affinis suspension culture (Seydel et al. 2007). We assessed cyclotide production levels in regenerated V. uliginosa plants showing different ploidy levels, and demonstrated that the quantity of cyclotides produced can be increased. The tendency of tetraploid regenerated V. uliginosa plants to synthesize more cyclotides suggests that applying chemicals such as colchicine to enhance polyploidization in culture can be advantageous. The tetraploid specimens with the highest cyclotide production can be used to establish higher-yielding suspension culture for bioreactors. Older plants with more differentiated tissues showed considerably lower cyclotide biosynthesis. These results indicate that cyclotide synthesis is more dependent on tissue development and genetic factors than on a direct response to stimuli, apparently supporting Mylne et al.’s (2010) assertion that these peptides are part of the plant’s innate defense system.
The in vitro system we developed for V. uliginosa opens an opportunity for cyclotide research as their expression can vary substantially in wild and cultivated plants. Apart from offering the prospect of generating the native cyclotides expressed by this rare species in large amounts, our study demonstrated that we can affect expression by the choice of somaclones and induce somaclonal variation by the growth conditions applied. In vitro culture is the standardized and controllable system we believe is needed to unravel the details of cyclotide biosynthesis. In fact, the methods developed in the present experiment were applied to study cyclotide biosynthesis regulation by plant growth regulators and to obtain highly productive suspension cultures (Slazak et al., in manuscript). Only common model plants (Arabidopsis thaliana, Nicotiana benthamiana, N. tabacum), have been used in functional studies of cyclotide biosynthesis (Gillon et al. 2008; Saska et al. 2007). None of them produce cyclotides naturally, so their yield of cyclic products is very low. In vitro cultures and micropropagation presents an attractive system for biotechnology applications (regeneration of transformed forms), or for the production of N15-labeled cyclotides, important for structural or pharmacokinetic studies. The incorporation of N15 into cyclotides has been demonstrated by Mylne and Craik (2008) in hydroponic cultivation of Oldenlandia affinis. Finally we note that violets are the main producers of both bracelet and Möbius cyclotides. With only one exception the difficulties presented by oxidative folding of bracelets have limited research involving chemical synthesis of these cyclotides (Aboye et al. 2008).
Conclusions
This work developed in vitro protocols for micropropagation of the endangered species V. uliginosa and a system for cyclotide biosynthesis studies in the Violeaceae. Molecular marker polymorphism and flow cytometric analyses indicated that in indirect organogenesis ploidy is the crucial factor to consider before reintroduction. The diploid plants are the ones to be reintroduced, and tetraploids can be used in suspension culture and bioreactors for their higher cyclotide biosynthesis. Indirect organogenesis via callus can be useful for creating large number of cyclotide-producing plants at higher than diploid level. Chemicals that enhance polyploidization, such as colchicine, should be included in further studies aimed at cyclotide production in vitro.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- 2C:
-
2C genome size
- ACN:
-
Acetonitrile
- AFLP:
-
Amplified fragment length polymorphism
- AUC:
-
Area under the curve
- DAPI:
-
4′,6-Amidino-2-phenylindole
- FA:
-
Formic acid
- IBA:
-
Indole-3-butyric acid
- KIN:
-
Kinetin
- MS:
-
Murashige and Skoog (1962) medium
- PBS:
-
Phosphate buffer
- PCoA:
-
Principal coordinate analysis
- PI:
-
Propidium iodide
- PVP:
-
Polyvinylpyrrolidone
- SPE:
-
Solid phase extraction
- TDZ:
-
Thidiazuron
References
Aboye TL, Clark RJ, Craik DJ, Göransson U (2008) Ultra-stable peptide scaffolds for protein engineering-synthesis and folding of the circular cystine knotted cyclotide cycloviolacin O2. ChemBioChem 9(1):103–113
Ahmad S, Garg M, Tajuddin Tamboli E, Abdin MZ, Ansari SH (2013) In vitro production of alkaloids: factors, approaches, challenges and prospects. Pharmacogn Rev 7:27–33
Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD et al (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep 30:108–160
Aversano R, Ercolano MR, Caruso I, Fasano C, Rosellini D, Carputo D (2012) Molecular tools for exploring polyploid genomes in plants. Intern J Mol Sci 13:10316–10335
Bairu MW, Aremu AO, Van Staden J (2011) Somaclonal variation in plants: causes and detection methods. Plant Growth Regul 63:147–173
Baryła J, Kuta E (2001) Viola uliginosa Besser, Fiołek bagienny. In: Kazmierczakowa R, Zarzycki K (eds) Polska czerwona księga roślin. Paprotniki i rośliny kwiatowe. Instytut Botaniki im. W. Szafera PAN, Instytut Ochrony Przyrody PAN, Kraków, pp 245–247
Bidwell SD, Pederick JW, Sommer-Knudsen J, Woodrow IE (2001) Micropropagation of the nickel hyperaccumulator, Hybanthus floribundus (Family Violaceae). Plant Cell Tissue Org 67:89–92
Bouque V, Bourgaud F, Nguyen C, Guckert A (1998) Production of daidzein by callus cultures of Psoralea species and comparison with plants. Plant Cell Tissue Org 53:35–40
Bourgaud F, Gravot A, Milesi S, Gontier E (2001) Production of plant secondary metabolites: a historical perspective. Plant Sci 161:839–851
Burman R, Gunasekera S, Strömstedt AA, Göransson U (2014) Chemistry and biology of cyclotides: circular plant peptides outside the box. J Nat Prod 77:724–736
Chalageri G, Babu UV (2012) In vitro plant regeneration via petiole callus of Viola patrinii and genetic fidelity assessment using RAPD markers. Turk J Bot 36:358–368
Cieślak E, Paul W, Ronikier M (2006) Low genetic diversity in the endangered population of Viola uliginosa in its locus classicus at Rząska near Cracow (Southern Poland) as revealed by AFLP markers. Acta Soc Bot Pol 75:245–251
Craik DJ (2012) Host-defense activities of cyclotides. Toxins (Basel) 4:139–156
Craik DJ, Conibear AC (2011) The chemistry of cyclotides. J Org Chem 76:4805–4817
Craik DJ, Daly NL, Bond T, Waine C (1999) Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J Mol Biol 294:1327–1336
D’Amato F (1991) Nuclear changes in cultured plant cells. Caryologia 44:217–224
Doležel J, Greilhuber J, Suda J (2007) Flow cytometry with plants: an overview. In: Doležel J, Greilhuber J, Suda J (eds) Flow cytometry with plant cells. Analysis of genes, chromosomes and genomes. Wiley-VCH Weinheim, Germany, pp 41–65
Dörnenburg H (2010) Cyclotide synthesis and supply: from plant to bioprocess. Biopolymers 94:602–610
Ehrich D (2006) AFLPDat: a collection of r functions for convenient handling of AFLP data. Mol Ecol Notes 6:603–604
Gärdenfors U (2005) Rödlistade arter i Sverige 2005.The 2005 Red list of Swedish species. Uppsala, ArtDatabanken, SLU, p 179
Gerlach SL, Göransson U, Kaas Q, David J, Craik DJ, Mondal D, Gruber CW (2013) A systematic approach to document cyclotide distribution in plant species from genomic, transcriptomic, and peptidomic analysis. Peptide Sci 100:433–437
Gillon AD, Saska I, Jennings CV, Guarino RF, Craik DJ, Anderson MA (2008) Biosynthesis of circular proteins in plants. Plant J. 53:505–515
Gruber CW, Elliott AG, Ireland DC, Delprete PG, Dessein S, Göransson U, Trabi M, Wang CK, Kinghorn AB, Robbrecht E, Craik DJ (2008) Distribution and evolution of circular miniproteins in flowering plants. Plant Cell 20:2471–2483
Ingelög T, Andersson R, Tjernberg M (1993) Red data book of the Baltic Region. Part 1. List of threatened vascular plants and vertebrates. Swedish Threatened Species Unit, Upsala & Institute of Biology, Riga, pp 25–68
Jedrzejczyk I, Sliwinska E (2010) Leaves and seeds as materials for flow cytometric estimation of the genome size of 11 Rosaceae woody species containing DNA-staining inhibitors. J Bot, ID 930895
Jennings CV, Rosengren KJ, Daly NL, Plan M, Stevens J, Scanlon MJ, Waine C et al (2005) Isolation, solution structure, and insecticidal activity of kalata B2, a circular protein with a twist: do Möbius strips exist in nature? Biochemistry 44:851–860
Korneck D, Schnittler M, Vollmer I (1996) Rote Liste der Farn- und Blütenpflatzen (Pteridophyta et Spermatophyta) Deutschlands. In: Rote liste gefährdeter Pflanzen Deutschlands. Schriftenreihe für Vegetationskunde 28:21–187
Kotiranta H, Uotila P, Sulkava S, Peltonen S (1998) Red data book of East Fennoscandia. Ministry of the Environment, Helsinki, pp 31–47
Krawczyk R, Nobis A, Nobis M, Cwenr A (2008) Is Viola uliginosa Besser critically endangered in Poland?—new data on the distribution of the species. Acta Soc Bot Pol 77:345–349
Kulkarni SS, Ravindra NS, Srinivas KV, Kulkarni RN (2012) A somaclonal variant of rose-scented geranium (Pelargonium spp.) with moderately high content of isomenthone in its essential oil. Nat Prod Commun 7:1223–1224
Larkin PJ, Scowcroft WR (1981) Somaclonal variation—a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214
Lee M, Phillips RL (1988) The chromosomal basis of somaclonal variation. Ann Rev Plant Physiol Plant Mol Biol 39:413–437
Lemontey C, Mousset-Déclas C, Munier-Jolain N, Boutin JP (2000) Maternal genotype influences pea seed size by controlling both mitotic activity during early embryogenesis and final endoreduplication level/cotyledon cell size in mature seed. J Exp Bot 51:167–175
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Mylne JS, Craik DJ (2008) 15 N Cyclotides by whole plant labeling. Biopolymers 90:575–580
Mylne JS, Wang CK, van der Weerden NL, Craik DJ (2010) Cyclotides are a component of the innate defense of Oldenlandia affinis. Biopolymers 94:635–646
Naeem M, Naveed I, Naqvi SMS, Mahmood T (2013) Standardization of tissue culture conditions and estimation of free scavenging activity in Viola odorata L. Pak J Bot 45:197–202
Neelakandan AK, Wang K (2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31:597–620
Nguyen GK, Lian Y, Pang EW, Nguyen PQ, Tran TD, Tam JP (2013) Discovery of linear cyclotides in monocot plant Panicum laxum of Poaceae family provides new insights into evolution and distribution of cyclotides in plants. J Biol Chem 288(5):3370–3380
Nybom H, Weising K, Rotter B (2014) DNA fingerprinting in botany: past, present, future. Invest Genet 5:1–35
Ochatt SJ, Patat-Ochatt EM, Moessner A (2011) Ploidy level determination within the context of in vitro breeding. Plant Cell Tissue Org 104:329–341
Pence VC (2013) In vitro methods and the challenge of exceptional species for target 8 of the global strategy for plant conservation. Ann Miss Bot Gard 99:214–220
Poth AG, Mylne JS, Grassl J, Lyons RE, Millar AH, Colgrave ML, Craik DJ (2012) Cyclotides associate with leaf vasculature and are the products of a novel precursor in petunia (Solanaceae). J Biol Chem 287(32):27033–27046
Prakash E, Sha Valli Khan PS, Sairam Reddy P, Rao KR (1999) Regeneration of plants from seed-derived callus of Hybanthus enneaspermus L. Muell., a rare ethnobotanical herb. Plant Cell Rep 18:873–878
Pränting M, Lööv C, Burman R, Göransson U, Andersson DI (2010) The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J Antimicrob Chemoth 65:1964–1971
R Development Core Team (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rewers M, Drouin J, Kisiala A, Sliwinska E, Cholewa E (2012) In vitro regenerated wetland sedge Eriophorum vaginatum L. is genetically stable. Acta Physi Plant 34:2197–2206
Rodriguez-Enriquez J, Dickinson HG, Grant-Downton RT (2011) MicroRNA misregulation: an overlooked factor generating somaclonal variation? Trend Plant Sci 16:242–248
Ronikier M, Cieślak E, Korbecka G (2008) High genetic dif-ferentiation in the alpine plan Campanula alpina Jacq. (Campanulaceae): evidence for glacial survival in several Carpathian regions and long-term isolation between the Carpathians and the Alps. Mol Ecol 17:1763–1775
Rosengren KJ, Daly NL, Plan MR, Waine C, Craik DJ (2003) Wists, knots, and rings in proteins. Structural definition of the cyclotide framework. J Biol Chem 278:8606–8616
Rout GR, Samantaray S, Das P (2000) In vitro manipulation and propagation of medicinal plants. Biotechnol Adv 18:91–120
Ryttäri T, Kettunen T (1997) Uhanalaiset kasvimme. Suomen ympäristökeskus. Kirjayhtymä Oy, Helsinki
Sarasan V, Cripps R, Ramsay MM, Atherton C, McMichen M, Prendergast G, Rowntree JK (2006) Conservation in vitro of threatened plants—progress in the past decade. In Vitro Cell Dev Biol Plant 42:206–214
Saska I, Gillon AD, Hatsugai N, Dietzgen RG, Hara-Nishimura I, Anderson MA, Craik DJ (2007) An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. J Biol Chem 282:29721–29728
Seydel P, Gruber CW, Craik DJ, Dörnenburg H (2007) Formation of cyclotides and variations in cyclotide expression in Oldenlandia affinis suspension cultures. Appl Microb Biotechnol 77:275–284
Singh SR, Dalal S, Singh R, Dhawan AK, Kalia RK (2013) Evaluation of genetic fidelity of in vitro raised plants of Dendrocalamus asper (Shult. & Shult. F.) Backer ex K. Heyne using DNA-based markers. Acta Physiol Plant 35:419–430
Soni M, Kaur R (2013) Rapid in vitro propagation, conservation and analysis of genetic stability of Viola pilosa. Physiol Mol Biol Plants. doi:10.1007/s12298-013-0200-8
Stoltze M, Pihl S (1998) Rødliste 1997 over planter og dyr I Danmark. København, pp 71–81
Thiem B, Sliwinska E (2003) Flow cytometric analysis of nuclear DNA content in cloudberry (Rubus chamaemorus L.) in vitro cultures. Plant Sci 164:129–134
Thiem B, Kikowska M, Krawczyk A, Więckowska B, Sliwinska E (2013) Phenolic acid and DNA contents of micropropagated Eryngium planum L. Plant Cell Tissue Org 114:197–206
Thorpe TA (2007) History of plant tissue culture. Mol Biotechnol 37:169–180
Vazquez AM (2001) Insight into somaclonal variation. Plant. Bios 135:57–62
Vos P, Hogers R, Bleeker M, Reijans M, Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wang J, Bao MZ (2007) Plant regeneration of pansy (Viola wittrockiana) ‘Caidie’ via petiole-derived callus. Scientia Hort 111:266–270
Wang Q-M, Wang L (2012) An evolutionary view of plant tissue culture: somaclonal variation and selection. Plant Cell Rep 31(9):1535–1547
Acknowledgments
This work was funded in part by the Dean of the Faculty of Biology and Earth Sciences of Jagiellonian University (project DS/MND/WBiNoZ/IB/4/2011 for A. Słomka, project DS/MND/WBiNoZ/IB/16/2011 for B. Slazak); an Ahlquists Stiftelse (Ahlquist Foundation) grant for research exchange at the Faculty of Pharmacy, Uppsala University; the Swedish Research Council (621-2007-5167); the Swedish Foundation for Strategic Research (F06-0058) (Ulf Göransson); and statutory funds of the Institute of Botany of the Polish Academy of Sciences (for M. Ronikier).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Slazak, B., Sliwinska, E., Saługa, M. et al. Micropropagation of Viola uliginosa (Violaceae) for endangered species conservation and for somaclonal variation-enhanced cyclotide biosynthesis. Plant Cell Tiss Organ Cult 120, 179–190 (2015). https://doi.org/10.1007/s11240-014-0592-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0592-3